Advanced Slurm Features
MonARCH uses the Slurm scheduler for running jobs. The home page for Slurm is http://slurm.schedmd.com/, and it is used in many computing systems, such as MASSIVE and VLSCI. Slurm is an open-source workload manager designed for Linux clusters of all sizes. It provides three key functions.
- It allocates exclusive and/or non-exclusive access to resources (computer nodes) to users for some duration of time so they can perform work.
- It provides a framework for starting, executing, and monitoring work (typically a parallel job) on a set of allocated nodes.
- It arbitrates contention for resources by managing a queue of pending work.
The following material will explain how users can use Slurm. At the bottom of the page there is a PBS, SGE comparison section. Slurm Glossary It is important to understand that some Slurm syntax have meanings which may differ from syntax in other batch or resource schedulers.
Glossary
Below is a summary of some Slurm concepts
| Term | Description |
|---|---|
| Task | A task under Slurm is a synonym for a process, and is often the number of MPI processes that are required |
| Success | A job completes and terminates well (with exit status 0) (cancelled jobs are not considered successful) |
| Socket | A socket contains one processor |
| Resource | A mix of CPUs, memory and time |
| Processor | A processor contains one or more cores |
| Partition | Slurm groups nodes into sets called partitions. Jobs are submitted to a partition to run. In other batch systems the term queue is used |
| Node | A node contains one or more sockets |
| Failure | Anything that lacks success |
| CPU | The term CPU is used to describe the smallest physical consumable, and for multi-core machines this will be the core. For multi-core |
| Core | A CPU core |
| Batch job | A chain of commands in a script file |
| Account | The term account is used to describe the entity to which used resources are charged to. This field is not used on the Monarch |
Useful Commands
| What | Slurm command | Comment |
|---|---|---|
| Job Submission | sbatch jobScript | Slurm directives in the jobs script can also be set by command line options for sbatch. |
| Check queue | squeue or aliases | You can also examine individual jobs, i.e. sq SQ squeue -j 792412 |
| Check cluster status | show_cluster | This is a nicely printed description of the current state of the machines in our cluster, built on top of the sinfo command |
| Deleting an existing job | scancel jobID | |
| Show job information | scontrol show job jobID | Also try show_job for nicely formatted output |
| Suspend a job | scontrol suspend jobID | |
| Resume a job | scontrol resume jobID | |
| Deleting parts of a job array | scancel jobID_5-10 |
More on Shell Commands
Users submit jobs to the MonARCH using Slurm commands called from the Unix shell (such as bash, or csh). Typically a user creates a batch submission script that specifies what computing resources they want from the cluster, as well as the commands to execute when the job is running. They then use sbatch <filename> to submit the job. Users can kill, pause and interrogate the jobs they are running. Here is a list of common commands:
sbatch
sbatch is used to submit a job script for later execution. The script will typically contain one or more srun commands to launch parallel tasks.
sbatch [options] job.script
scancel
scancel deletes a job from the queue, or stops it running.
scancel jobID1 jobID2
scancel --name=[job name]
scancel --user=[user]
sinfo
sinfo reports the state of partitions and nodes managed by Slurm. It has a wide variety of filtering, sorting, and formatting options.
sinfo [options]
Example:
sinfo
PARTITION AVAIL TIMELIMIT NODES STATE NODELIST
comp* up 7-00:00:00 2 mix gp[00-01]
comp* up 7-00:00:00 23 idle gp[02-05],hc00,hs[00-05],mi[00-11]
squeue
squeue reports the state of jobs or job steps. It has a wide variety of filtering, sorting, and formatting options. By default, it reports the running jobs in priority order and then the pending jobs in priority order.
#Print information only about job step 65552.1:
squeue --steps 65552.1
STEPID NAME PARTITION USER TIME_USE NODELIST(REASON)
65552.1 test2 debug alice 12:49 dev[1-4]
We also have set up alises to squeue that prints more information for them.
| Alias | Maps to |
|---|---|
| sq | squeue -u userid |
| SQ | squeue -o %.18i %.8P %.6a %.15j %.8u %8Q %.8T %.10M %.4c %.4C %.12l %.12L %.6D %.16S %.16V %R |
SQ prints more information on the jobs, for all users and can be used like:
SQ -u myUserName
Some squeue options of interest. See man squeue for more information.
| squeue option | Meaning |
|---|---|
| --array | Job arrays are displayed one element per line |
| --jobs=JobList | Comma separated list of Job IDs to display |
| --long | Display output in long format |
| --name=NameList | Filter results based on job name |
| --partition=PartitionList | Comma separated list of partitions to display |
| --user=User | Display results based on the listed user name |
scontrol
scontrol reports or modify details of a currently running job. (Use sacct to view details on finished jobs)
scontrol show job 71701 #report details of job whose jobID is 71701
scontrol show jobid -dd 71701 # report more details on this job, including the submission script
scontrol hold 71701 #hold a job, prevents the job being scheduled for execution
sctonrol release 71701 #release a job that was previously held manually
sinteractive
It is possible to run a job as an interactive session using 'sinteractive '. The program hangs until the session is scheduled to run, and then the user is logged into the compute node. Exiting the shell (or logging out) ends the session and the user is returned to the original node.
sinteractive
Waiting for JOBID 25075 to start
Warning: Permanently added 'm2001,172.19.1.1' (RSA) to the list of known hosts.
$hostname
m2001
$exit
[screen is terminating]
Connection to m2001 closed.
sacct
The command sacct shows metrics from past jobs.
sacct -l -j jobID
sstat
The command sstat shows metrics from currently running jobs when given a job number. Note, you need to launch jobs with srun to get this information
Help on shell commands
Users have several ways of getting information on shell commands.
- The commands have man pages (via the unix manual). e.g. man sbatch
- The commands have built-in help options. e.g.
sbatch --help
sbatch --usage. - There are online manuals and information pages
Most commands have options in two formats:
- single letter e.g. -N 1
- verbose e.g. --nodes=1
Note the double dash -- in the verbose format. A non-zero exit code indicates failure in a command.
Some default behaviours:
- Slurm processes launched with srun are not run under a shell, so none of the following are executed:
- ~/.profile
- ~/.baschrc
- ~/.login
- Slurm exports user environment by default (or --export=NONE)
- Slurm runs in the current directory (no need to cd $PBS_O_WORKDIR)
- Slurm combines stdout and stderr and outputs directly (and naming is different). The Slurm stdout /stderr file will be appended,not overwritten (if it exists)
- Slurm is case insensitive (e.g. project names are lower case)
Batch Scripts
A job script has a header section which specifies the resources that are required to run the job as well as the commands that must be executed. An example script is shown below.
!/bin/env bash
#SBATCH --job-name=example
#SBATCH --time=01:00:00
#SBATCH --ntasks=32
#SBATCH --ntasks-per-node=16
#SBATCH --cpus-per-task=1
#SBATCH --mem=2000
module load intel
uname -a
srun uname -a
Here are some of the Slurm directives you can use in a batch script. man sbatch will give you more information.
| Slurm directive | Description |
|---|---|
| --job-name=[job name] | The job name for the allocation, defaults to the script name. |
| --partition=[partition name] | Request an allocation on the specified partition. If not specified jobs will be submitted to the default patition. |
| --time=[time spec] | The total walltime for the job allocation. |
| --array=[job spec] | Submit a job array with the defined indices. |
| --dependency=[dependency | Specify a job dependency list] |
| --nodes=[total nodes] | Specify the total number of nodes. |
| --ntasks=[total tasks] | Specify the total number of tasks. |
| --ntasks-per-node=[ntasks] | Specify the number of tasks per node. |
| --cpus-per-task=[ncpus] | Specify the number of CPUs per task. |
| --ntasks-per-core=[ntasks] | Specify the number of tasks per CPU core. |
| --export=[variable|ALL|NONE] | Specify what environment variables to export. NOTE: Slurm will copy the entire environment from the shell where a job is submitted from. This may break existing batch scripts that require a different environment than say a login environment. To guard against this --export=NONE can be specified for each batch script. |
Comparing PBS/SGE instructions with Slurm
This section provides a brief comparison of PBS, SGE and Slurm input parameters. Please note that in some cases there is no direct equivalent between the different systems.
Basic Job commands
| Comment | MASIVE PBS | SGE | Slurm |
|---|---|---|---|
| Give the job a name. | #PBS -N JobName | #$ -N JobName | #SBATCH --job-name=JobName #SBATCH -J JobName Note: The job name appears in the queue list, but is not used to name the output files (opposite behavior to PBS, SGE) |
| Redirect standard output of job to this file. | #PBS -o path | #$ -o path | #SBATCH --output=path/file-%j.ext1 #SBATCH -o path/file-%j.ext1 Note: %j is replaced by the job number |
| Redirect standard error of job to this file. | #PBS -e path | #$ -e path | #SBATCH --error=path/file-%j.ext2 #SBATCH -e path/file-%j.ext2 Note: %j is replaced by the job number. |
Commands to specify accounts, queues and working directories.
| Comment | MASSIVE PBS | SGE | Slurm |
|---|---|---|---|
| Account to charge quota. (if so set up) | #PBS -A AccountName | #SBATCH --account=AccountName #SBATCH -A AccountName | |
| Walltime | #PBS -l walltime=2:23:59:59 | # -l h_rt=hh:mm:ss e.g. # -l h_rt=96:00:00 | #SBATCH --time=2-23:59:59 #SBATCH -t 2-23:59:59 Note '-' between day(s) and hours for Slurm. |
| Change to the directory that the script was launched from | cd $PBS_O_WORKDIR | #$ -cwd | This is the default for Slurm. |
| Specify a queue (partition) | #PBS -q batch | #$ -cwd | #SBATCH --partition=main #SBATCH -p main Note: In Slurm a queue is called a partition, and the default is 'batch'. |
Instructions to request nodes, sockets and cores.
| Comment | MASSIVE PBS | SGE | Slurm |
|---|---|---|---|
| The number of compute cores to ask for | #PBS -l nodes=1:ppn=12 Asking for 12 CPU cores, which is all the cores on a MASSIVE node. You could put "nodes=1" for a single CPU core job or "nodes=1:ppn=4" to get four cpu cores on the one node (typically for multithreaded, smp or openMP jobs) | #$ -pe smp 12 #$ -pe orte_adv 12 MCC SGE did not implement running jobs across machines, due to limitations of the interconnection hardware | #SBATCH --nodes=1 --ntasks=12 or #SBATCH -N1 -n12 --ntasks is not used in isolation but combined with other commands such as --nodes=1 |
| The number of tasks per socket | --ntasks-per-socket= Request the maximum ntasks be invoked on each socket. Meant to be used with the --ntasks option. Related to --ntasks-per-node except at the socket level instead of the node level | ||
| Cores per task (for use with openMP) | --cpus-per-task=ncpus or -c ncpus Request that ncpus be allocated per process. The default is one CPU per process. | ||
| Specify per core memory. | #PBS -l pmem=4000MB Specifies how much memory you need per CPU core (1000MB if not specified) | No equivalent. SGE uses memory/process | --mem-per-cpu=24576 or --mem=24576 Slurm default unit is MB. |
Commands to notify user of job progress.
| Comment | MASSIVE PBS | SGE | Slurm |
|---|---|---|---|
| Send email notification when: job fails. | #PBS -m a | #$ -m a | #SBATCH --mail-type=FAIL |
| Send email notification when: job fails. | #PBS -m b | #$ -m b | #SBATCH --mail-type=BEGIN |
| Send email notification when: job stops. | #PBS -m e | #$ -m e | #SBATCH --mail-type=END |
| e-mail address to send information to | #PBS -M name@email.address | #$ -M name@email.address | #SBATCH --mail-user=name@email.address |
Examples of advanced Slurm features
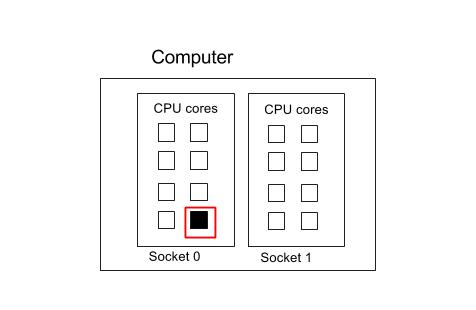
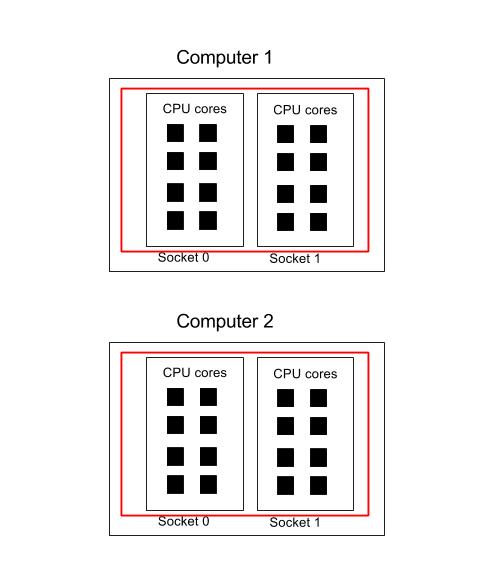
This section focuses on how to specify the different CPU resources you need. See below for a block diagram of a typical compute node. This consists of a mother board with two CPU sockets, and in each socket is a 8-core CPU. (Note, not all machines are like this of course. The motherboard may have more CPU sockets, and their may be more cores/CPU in each processor).
A process (or task) is identified by a thick red line. If a core is used by a thread or process, it is coloured black. So in the diagram below we have one process that has one thread.
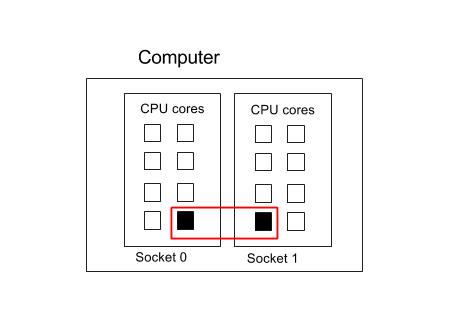
In the following example, there is one process that is using two threads. These threads have been allocated to two different sockets.
Serial Example
This section describes how to specify a serial job - namely one that uses only one core on one processor.
#!/bin/env bash
#SBATCH --ntasks=1
#SBATCH --ntasks-per-node=1
#SBATCH --cpus-per-task=1
serialProg.exe
OpenMP
OpenMP is a thread-based technology based on shared memory systems, i.e. OpenMP programs only run on one computer. It is characterized by having one process running multiple threads.
Slurm does not set OMP_NUM_THREADS in the environment of a job. Users should manually add this to their batch scripts, which is normally the same as that specified with --cpus-per-task
To compile your code with OpenMP
| Compiler | Option To Use When Compiling |
|---|---|
| gcc | -fopenmp |
| icc | -openmp |
Note that the command below explicitly states the NUMA configuration of the processes. You need not specify all the commands if this is not important.
8 Cores on 1 Socket
Suppose we had a single process that we wanted to run on all the cores on one socket.
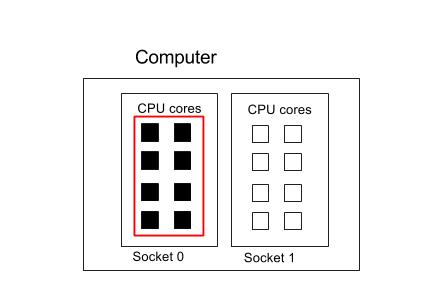
#!/bin/env bash
#SBATCH --nodes=1
#SBATCH --ntasks=1
#SBATCH --cpus-per-task=8
#SBATCH --ntasks-per-socket=1
# Set OMP_NUM_THREADS to the same value as --cpus-per-task=8
export OMP_NUM_THREADS=8
./openmp_program.exe
8 Cores spread across 2 Sockets
Now suppose we wanted to run the same job but with four cores per socket
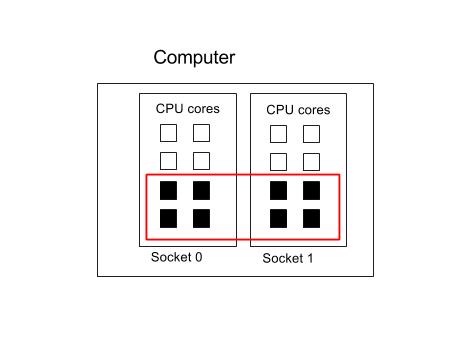
#!/bin/env bash
#SBATCH --nodes=1
#SBATCH --ntasks=1
#SBATCH --cpus-per-task=8
#SBATCH --cores-per-socket=4
# Set OMP_NUM_THREADS to the same value as --cpus-per-task=8
export OMP_NUM_THREADS=8
openmp_program.exe
MPI Examples
This section contains a number of MPI examples to illustrate their usage with Slurm. MPI processes can be started with the srun command, or the traditional mpirun or mpiexe. There is no need to specify the number of process to run (-np) as this is automatically read in from Slurm environment variables.
For MonARCH-only:
- See MPI on MonARCH for info on the best use of MPI on our system.
- Instead of hard-coding the number of processors into the mpirun command line, i.e. 32, you may want to use a Slurm environment variable, SLURM_NTASKS, to make the script more generic. (Note that if you use threading or openMP you will need to modify the parameter. See below)
Note that the command below explicitly states the NUMA configuration of the processes. You need not specify all the commands if this is not important.
Four processes in one socket
Please note that in this example, we are constraining processes to run inside a single socket. You can leave out the --ntasks-per-socket command to let the Operating System choose where to put the processes.
Suppose we want to use 4 cores in one socket for our four MPI processes, with each process running on one core each.
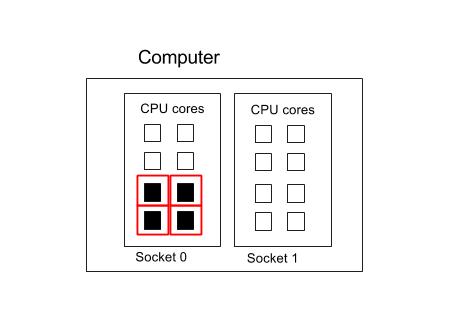
#!/bin/env bash
#SBATCH --ntasks=4
#SBATCH --ntasks-per-node=4
#SBATCH --cpus-per-task=1
#SBATCH --ntasks-per-socket=4
# you can also explicitly state
# #SBATCH --nodes=1
# but this is implied already from the parameters we have specified.
module load openmpi
srun myMpiProg.exe # or mpirun mpiProg.ex
or
FLAGS="--mca btl_tcp_if_exclude virbr0"
mpirun -n $SLURM_NTASKS $FLAGS myMpiProg.exe
Four processes spread across 2 sockets
Please note that in this example, we are constraining processes to run inside two sockets. You can leave out the --ntasks-per-socket command to let the Operating System choose where to put the processes.
Suppose we want to use 4 cores spread across two socket for our four MPI processes, with each process running on one core each. (This configuration may minimize memory IO contention).
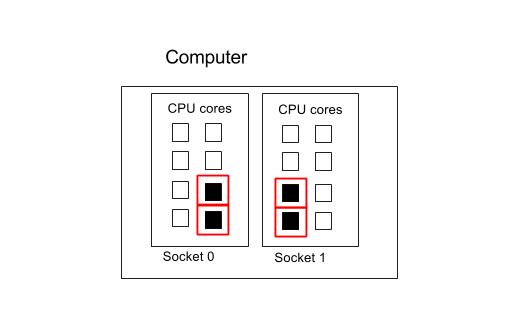
#!/bin/env bash
#SBATCH --ntasks=4
#SBATCH --ntasks-per-socket=2
#SBATCH --cpus-per-task=1
# you can also explicitly state
# #SBATCH --nodes=1
# but this is implied already from the parameters we have specified.
module load openmpi
srun mpiProg.exe # or mpirun mpiProg.exe
or
FLAGS="--mca btl_tcp_if_exclude virbr0"
mpirun -n $SLURM_NTASKS $FLAGS myMpiProg.exe
Using all the cores in one computer
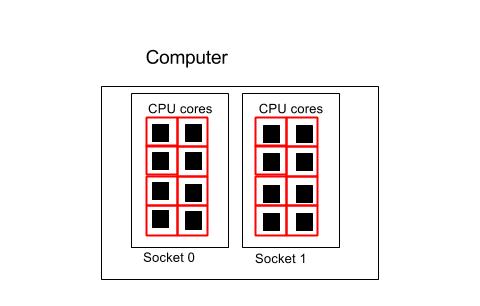
#!/bin/env bash
#SBATCH --ntasks=16
#SBATCH --ntasks-per-socket=8
#SBATCH --cpus-per-task=1
# you can also explicitly state
# #SBATCH --nodes=1
# but this is implied already from the parameters we have specified.
module load openmpi
srun myMpiProg.exe #or mpirun mpiProg.exe
or
FLAGS="--mca btl_tcp_if_exclude virbr0"
mpirun -n $SLURM_NTASKS $FLAGS myMpiProg.exe
Using all the cores in 2 computers
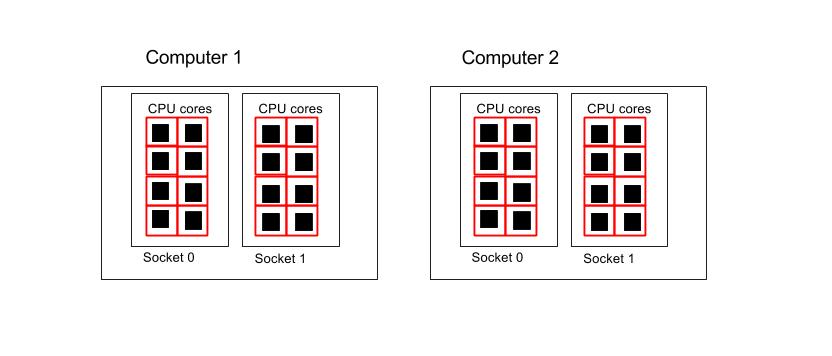
#!/bin/env bash
#SBATCH --ntasks=32
#SBATCH --cpus-per-task=1
# you can also explicitly state
# #SBATCH --nodes=2
# but this is implied already from the parameters we have specified.
module load openmpi
srun mpiProg.exe
or
FLAGS="--mca btl_tcp_if_exclude virbr0"
mpirun -n $SLURM_NTASKS $FLAGS myMpiProg.exe
Hybrid OpenMP/MPI Jobs
It is possible to run MPI tasks which are in turn multi-threaded, e.g. with OpenMPI. Here are some examples to assist you.
Two nodes with 1 MPI process per node and 16 OpenMP threads each
#!/bin/env bash
#SBATCH --job-name=MPIOpenMP
#SBATCH --ntasks=2
#SBATCH --ntasks-per-node=1
#SBATCH --cpus-per-task=16
#SBATCH --time=00:05:00
export OMP_NUM_THREADS=16
srun ./HybrudMpiOpenMPI.exe
Using GPUs
This is how to invoke GPUs on Slurm on MonARCH.
One GPU and one core.
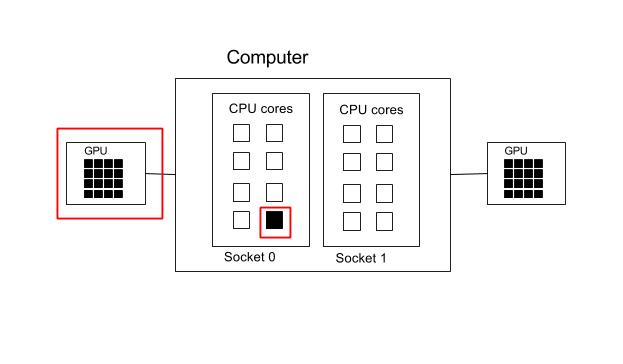
#!/bin/env bash
#SBATCH --nodes=1
#SBATCH --ntasks=1
#SBATCH --cpus-per-task=1
#SBATCH --ntasks-per-socket=1
#SBATCH --gres=gpu:K80:1
./gpu_program.exe
Two GPU and all cores.
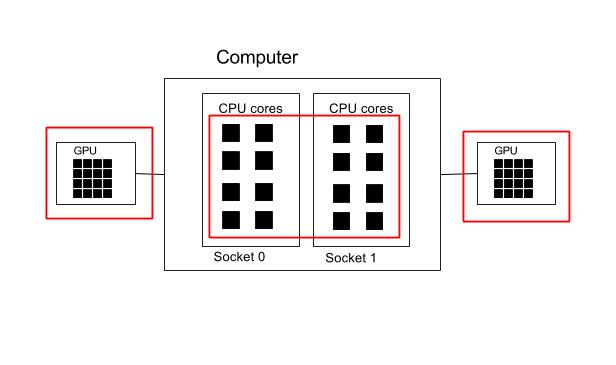
#!/bin/env bash
#SBATCH --nodes=1
#SBATCH --ntasks=1
#SBATCH --cpus-per-task=16
#SBATCH --gres=gpu:K80:2
./gpu_program.exe
Slurm: Array Jobs
It is possible to submit array jobs, that are useful for running parametric tasks. The command
#SBATCH --array=1-300
specifies that 300 jobs are submitted to the queue, and each one has a unique identifier specified in the environment variable SLURM_ARRAY_TASK_ID (in this case ranging from 1 to 300).
#!/bin/env bash
#SBATCH --job-name=sample_array
#SBATCH --time=10:00:00
#SBATCH --mem=4000
# make 300 different jobs from this one script!
#SBATCH --array=1-300
#SBATCH --output=job.out
module load modulefile
myExe.exe ${SLURM_ARRAY_TASK_ID} # equivalent to SGE's ${SGE_TASK_ID}
There are pre-configured limits to how many array jobs that you can submit in a single request.
scontrol show config | grep MaxArraySize
MaxArraySize = 5000
So in MonARCH, you can only submit 5000 array jobs per submission. This parameter also constrains the maximum index size to 5000.
If you want to exceed the index size, you can use arithmetic inside your Slurm submission script. For example, to increase the index so that you scan from 5001 to 10000 you can go
#SBATCH --array=1-5000
x=5000
new_index=$((${SLURM_ARRAY_TASK_ID} + $x))
#new_index will then range from 5001 to 10000
Email Notification
Slurm can email users with information on their job. This is enabled with the following flags.
#!/bin/env bash
#SBATCH --mail-type=FAIL,BEGIN,END
#SBATCH --mail-user=researcher@monash.edu
See man sbatch for all options assoicated with the email alerts.